Lysosomal storage disorders: A care journey with many challenges
Published Sep 25, 2021 • By Clémence Arnaud
Lysosomal storage disorders are a subgroup of diseases included in the rare diseases classification. According to a study by the Rare Disease Observatory, 21% of the people surveyed waited more than 6 years before obtaining a diagnosis of their disease.
What are lysosomal storage disorders? Are there treatments? How to live with a lysosomal storage disorder?
We explain it all in our article!

What are lysosomal storage disorders?
Lysosomal storage disorders (LSDs) are a group of about fifty, rare and inherited metabolic diseases. These are genetic diseases that impact cell function and more specifically lysosomes.
Lysosomes: Key subcellular organelles
Lysosomes are a membrane-bound organelles in our body's cells that clean the cell of the waste products they produce during their function.
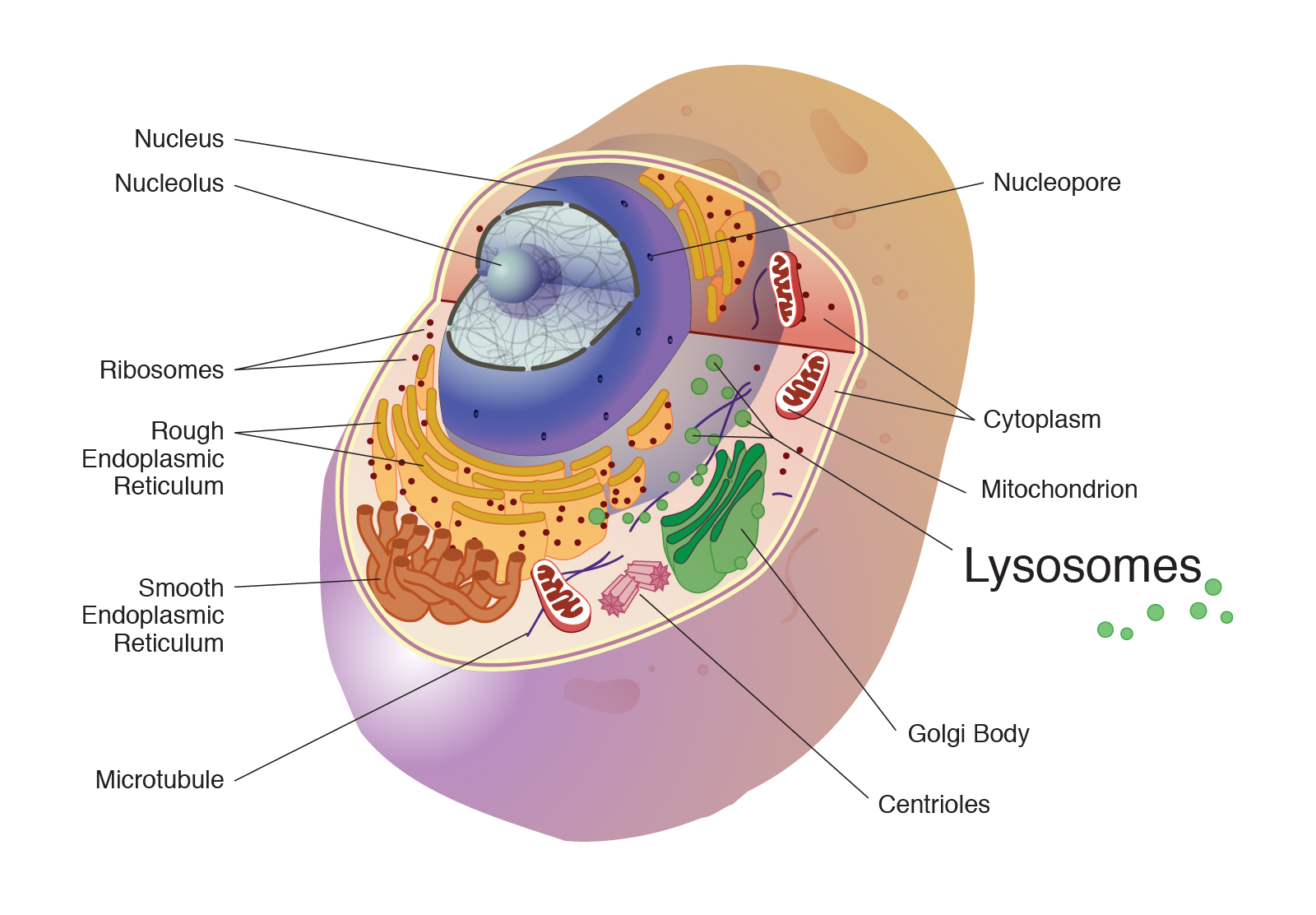 Source: NIH National Human Genome Research Institute
Source: NIH National Human Genome Research Institute
Lysosomes contain many digestive enzymes that allow them to break down and recycle excess or worn-out cell parts. If one of these enzymes is defective due to a mutation, the waste material accumulates within the cell, eventually killing it. In other words, when the lysosome does not function normally, excess products destined for breakdown and recycling are stored in the cell.
Total absence of enzymes or enzyme deficiencies lead to an excess of lysosomes, which in turn cause cell dysfunction. It is this phenomenon that occurs mainly in the case of lysosomal storage disorders.
In other cases, it is the deficit or absence of transport proteins that cause lysosomal overload. The lysosome is no longer able to evacuate waste, which causes cell dysfunction.
Depending on the type of mutation and the gene that is mutated, there will be different consequences for the individual depending on the enzyme that is impacted.
Classification of lysosomal storage disorders
LSDs typically classed into subgroups according to the main stored material that will be accumulated in the lysosome:
- Lipid storage disorders: accumulation of fats or lipids in the lysosomes (Fabry disease, etc.)
- Sphingolipidoses, including Gaucher's and Niemann-Pick diseases
- Gangliosidosis, including Tay-Sachs disease
- Leukodystrophies, including metachromatic leukodystrophy (MLD), Krabbe disease, X-Linked adrenoleukodystrophy (ALD), Canavan disease, and Alexander disease
- Mucopolysaccharidoses, including Hunter syndrome and Hurler disease
- Glycoprotein storage disorders (glycoproteinosis), including Pompe disease, Alpha-Mannosidosis, etc.
- Lysosomal transfer abnormalities, including cystinosis and Salla disease
- Neuronal ceroid lipofuscinosis (NCL), including CLN3 disease
- Other lysosomal storage disorders: Pycnodysostosis, Papillon-Lefèvre syndrome, etc.
Transmission of lysosomal storage disorders
Most lysosomal storage disorders are inherited in an autosomal recessive manner.
Most lysosomal diseases are transmitted to the next generation based on the principal of autosomal recessive transmission.
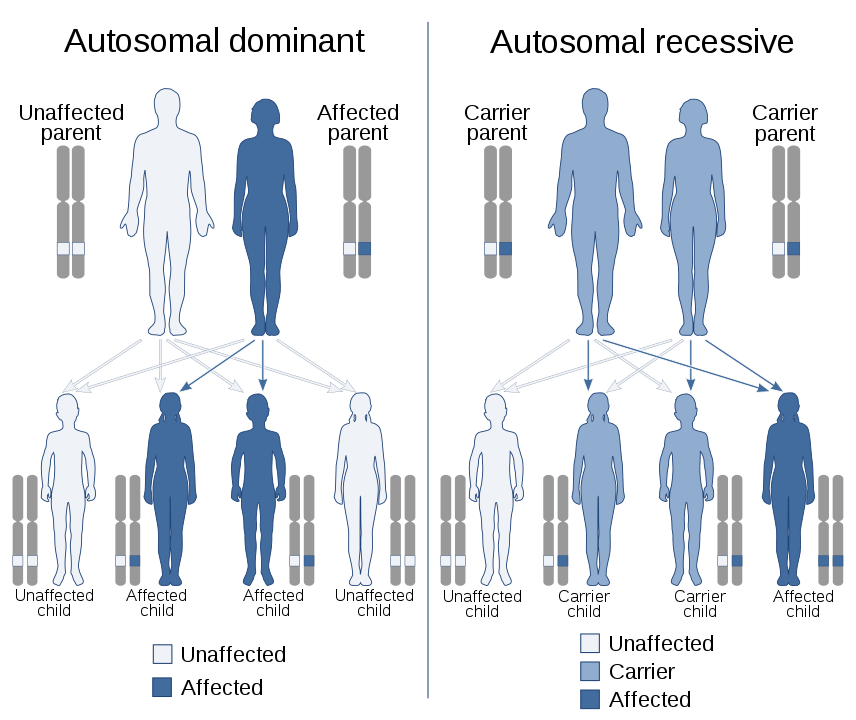
Source: Wikimedia Commons
The most common case is illustrated in the image above. Both parents have only one part of their gene, called an allele, carrying the mutation. For an individual to have symptoms of the condition, both parts of the gene must be affected by the mutation. If neither parent has symptoms, they are said to be carriers. There is then a risk of transmission to their offspring:
- In 25% of cases, the child will have no part of the mutated gene, and will be unaffected.
- In 50% of cases, the child will have only one part of the mutated gene, and will be a carrier.
- In 25% of cases, the child will carry both parts of the mutated gene and will be affected by the disease.
Only the Hunter, Danon and Fabry lysosomal storage diseases are linked to the X chromosome.
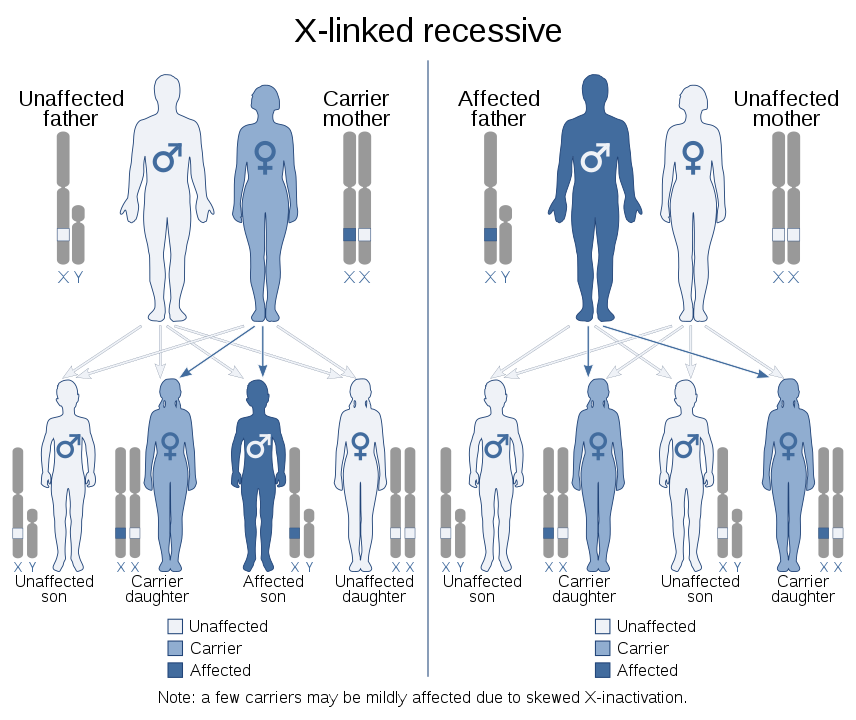
Source: Wikimedia Commons
Both X chromosomes must be affected by the mutation for the person to express the symptoms of the disease. Women have one X and one Y chromosome, so these conditions mostly affect men and very rarely women.
You can find more information on the genetic mutations in our article: What is a genetic disorder?
What are the treatment options for lysosomal storage disorders?
The care pathway
The care pathway for patients with rare diseases is often very complicated and burdensome for patients and their families. Clinical guidelines and care protocols have been put in place to optimize the care of these patients. They rely on specific structures with expertise in the management of rare diseases. Regional specialist clinical centers exist with the aim of reducing diagnostic errancy, i.e. the time it takes before a patient is diagnosed. Another objective is to facilitate patient care by minimizing the impact of treatments and medical appointments on the daily lives of patients and their families.
Much work still needs to be done to increase medical expertise in rare disease and increase patient access to specialist in rare diseases.
Available treatment options
A number of therapies are available for lysosomal storage disorders, you can read more about them in detail on the National Organization for Rare Diseases website.
These are not treatments that will cure the patient but that will slow down the progression and limit the symptoms of the disease.
One example is enzyme replacement therapy (ERT), the aim of which is to provide the patient with the missing enzyme to enable the cells to function. Substrate reduction therapy will limit the production of the molecule that cannot be broken down by the lysosomes because of the missing enzyme.
Many other avenues such as gene therapy are being developed to find a treatment that will hopefully lead to a cure for patients with LSDs.
Going beyond drug treatment:
The impact of these diseases on patients' lives is very significant. Patients' life expectancy can be impacted to a greater or lesser extent by the disease, especially when there is no treatment. About 50% of patients or parents of children with rare diseases have admitted to having stopped working and the same percentage feel isolated. Because of the impact of the disease on many organs, care must be adapted in the best possible way to each patient.
Therapeutic education courses are being developed. The aim of therapeutic education is to improve patients' quality of life and to enable them to live better and to better understand their condition. These programs are carried out with a team of health professionals: doctors, nurses, pharmacists, etc. In addition, patient associations or long-time patients may also be involved.
Was this article helpful to you?
Give it a like and share your thoughts and questions with the community in the comments below!
Take care!
 Facebook
Facebook Twitter
TwitterSources:
- National Organisation for Rare Disorders
- Lysosome, NIH National Human Genome Research Institute
- National Gaucher Foundation
- Orphanet - Transmission des maladies génétiques
- Ministère des solidarités et de la santé - L'offre de soins
- Maladies rares info services - L'observatoire des maladies rares 2015
- HAS - Protocoles nationaux de diagnostic et de soins


